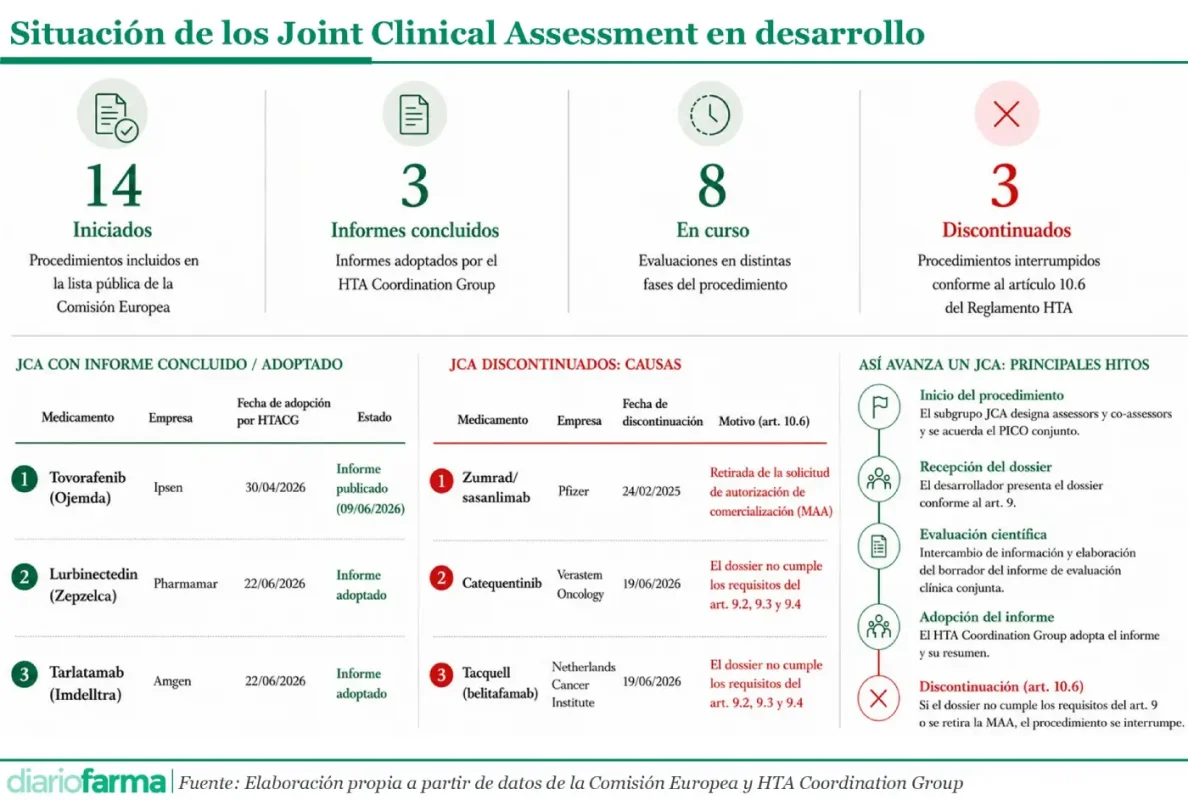
La evaluación clínica conjunta europea empieza a mostrar sus primeras señales de madurez. Tras la publicación del primer informe de JCA correspondiente a tovorafenib (Ojemda), el HTA Coordination Group ha adoptado otros dos informes, relativos a lurbinectedina (Zepzelca), de PharmaMar, y tarlatamab (Imdylltra), de Amgen, ambos en cáncer de pulmón microcítico en estadio extendido.
La novedad no reside ya en la existencia del primer informe, sino en que el procedimiento continúa su avance, con nuevos expedientes concluidos. Según la información disponible, estos dos nuevos informes han sido adoptados por el HTA Coordination Group y remitidos a la Comisión Europea, por lo que están pendientes del visto bueno procedimental de la Comisión antes de su publicación final.
La lista pública de la Comisión Europea, actualizada el 24 de junio de 2026, permite observar ya una primera fotografía del sistema: 14 procedimientos iniciados, tres informes concluidos o adoptados, ocho evaluaciones en curso y tres procedimientos discontinuados.
Tres procedimientos discontinuados
Junto al avance de los informes concluidos, la otra novedad relevante es la existencia de tres JCA discontinuados: Zumrad/sasanlimab, catequentinib y Tacquell/belitafamab.
Las causas no son homogéneas. En el caso de Zumrad/sasanlimab, la discontinuación está vinculada a la retirada de la solicitud de autorización de comercialización ante la EMA. Al desaparecer el procedimiento regulatorio de referencia, el JCA pierde su objeto.
Los casos de catequentinib y Tacquell tienen una lectura diferente. Según la información recogida en la lista pública y en análisis sectoriales, la discontinuación se relaciona con el incumplimiento de los requisitos del dossier previstos en el artículo 9 del Reglamento HTA, tras el procedimiento contemplado en el artículo 10.6.
Este punto es especialmente relevante porque muestra que el JCA no es un trámite automático. El desarrollador debe presentar un dossier completo, trazable y suficiente para responder al alcance de evaluación definido. Si, tras una segunda solicitud de información, el dossier no se presenta a tiempo o no cumple los requisitos exigidos, el Grupo de Coordinación debe discontinuar la evaluación.
Qué significa para la industria
La primera lectura para las compañías es clara: la preparación del JCA debe empezar mucho antes de la autorización europea. No basta con disponer de evidencia clínica; será necesario presentarla de forma estructurada, reproducible y alineada con los PICO definidos en el procedimiento.
Las tres discontinuaciones no deben interpretarse de forma automática como una valoración clínica negativa de los medicamentos. En un caso, el motivo es regulatorio; en los otros dos, el problema parece estar en la suficiencia y completitud del dossier. Pero, en conjunto, los tres expedientes envían una señal relevante: el nuevo marco europeo tendrá consecuencias procedimentales reales.
Un sistema que empieza a consolidarse
El balance actual muestra un sistema todavía inicial, pero ya operativo. Hay informes publicados o adoptados, evaluaciones en curso y procedimientos interrumpidos. Es decir, el JCA ha pasado de ser una previsión normativa a convertirse en un proceso con efectos concretos para compañías, agencias evaluadoras y sistemas nacionales.
Los próximos meses permitirán comprobar si la publicación de lurbinectedina y tarlatamab confirma una cadencia regular de informes y si las discontinuaciones observadas son casos puntuales o una advertencia sobre la preparación real de la industria ante el nuevo Reglamento HTA.








 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


