La Agencia Española de Medicamentos y Productos Sanitarios (Aemps) ha publicado una nota informativa con recomendaciones a la industria farmacéutica para la implementación de los dispositivos de seguridad en los medicamentos de uso humano, de cara al cumplimiento de las obligaciones que establece el Reglamento Delegado UE/2016/161, las cuales serán aplicables a partir del 9 de febrero de 2019. En este sentido, la agencia reconoce que dicha aplicación "obliga a decisiones que deberán estar incluidas en un desarrollo normativo". No obstante, y para que se vayan dando pasos que permitan ir cumpliendo plazos, han decidido hacer públicas estas recomendaciones.
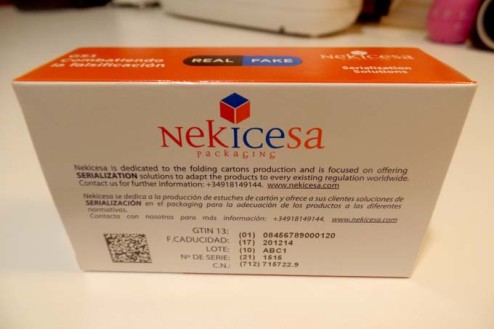
El primer aspecto al que se refiere la nota es a la inclusión del Código Nacional (CN) en el identificador único (IU), una posibilidad recogida en el Reglamento Delegado. La Agencia señala que, tras la discusión con las autoridades sanitarias competentes en prestación farmacéutica (las comunidades autónomas, principalmente), "se ha considerado procedente la inclusión de dicho Código como requisito adicional a la composición del identificador único". "El CN deberá quedar incorporado como uno de los elementos del IU, manteniéndose en el ángulo superior derecho de la cara principal del embalaje exterior o en la esquina superior derecha de la blue box", explican. Por el contrario, no será necesario imprimirlo de forma adyacente al Datamatrix de forma adicional.
Finalmente, la Aemps ha decidido que el CN quedará incorporado a la información contenida en el Datamatrix, dejando abierta la posibilidad de que se incorpore al código NTIN (National Trade Item Number) o como información adicional en el caso de que los medicamentos vayan codificados con el GTIN (Global Trade Item Number). En varias ocasiones, expertos y responsables de la industria habían opinado que era recomendable la migración total al GTIN, para converger con otros países en el sistema de codificación, pero por el momento la autoridad regulatoria ha optado por un modelo en convivencia.
La Agencia acepta que la incorporación progresiva de los Datamatrix hasta 2019 va a implicar la coexistencia con el código de barras actual incluido en el cupón precinto para el reembolso. En este sentido, la Aemps avanza que la utilización del nuevo sistema de codificación para el sistema de prestación farmacéutica exige "la publicación de una normativa específica".
Una vez incorporados los identificadores únicos en los envases y estén estos liberados para su venta o distribución, serán los titulares de comercialización los que garanticen la correcta lectura de todos sus elementos, para lo cual deberán haberlos cargado ya en el repositorio. Los que hayan sido liberados antes del 9 de febrero de 2019 podrán comercializarse, distribuirse y dispensarse hasta su fecha de caducidad.
Trámites para modificar el etiquetado
Además de la información sobre las características del IU, la Aemps ha explicado el procedimiento que habrá que seguir para la modificación del etiquetado para incorporar el dispositivo de seguridad. En este sentido, distingue, por un lado, aquellos casos en los que no se requiere presentar variación, como cuando la inclusión se haga en un espacio no utilizado del envase; sólo requiera la eliminación de información repetida, o requiera la reubicación de contenido sin afectar a la legibilidad. En esos casos, las compañías sí tendrán que cargar las maquetas de diseño actualizadas en la aplicación RAEFAR, siguiendo el manual de instrucciones que se publicará a tal efecto, así como garantizar la actualización del expediente electrónico del producto, estando la operación estará exenta del pago de tasas.
En cambio, los laboratorios tendrán que solicitar variación de tipo IB C.I.z, correspondiente al cambio de diseño, cuando la inclusión suponga eliminar contenido de información autorizada por la Aemps, una reorganización del cartonaje que afecte a la legibilidad, así como los cambios que no tengan que ver con la implementación del dispositivo. Como las anteriores, estas empresas tendrán que solicitar la notificación de dispositivos de seguridad, para verificar el cumplimiento de la medida.
Otro trámite obligatorio será el de la solicitud de una modificación del módulo 3 de calidad para la incoporación de los dispositivos contramanipulaciones en los medicamentos que carezcan de embalaje exterior, que también requerirá la notificación anteriormente citada.
Para aquellos medicamentos autorizados por procedimiento europeo, las compañías deberán seguir los mismos procedimientos establecidos para los autorizados por procedimiento nacional, presentando notificación de dispositivo de seguridad y con exención de tasas, para las que no modifiquen el envase, y siguiendo el procedimiento trazado para las que tienen que solicitar variación, con el calendario y el pago de tasas correspondiente. Una vez autorizado a nivel europeo, tendrán que notificar los nuevos materiales autorizados para asegurar la contabilidad del cumplimiento de la medida.

















 César Hernández, director general de Cartera y Farmacia del Ministerio de Sanidad:
César Hernández, director general de Cartera y Farmacia del Ministerio de Sanidad:  Kilian Sánchez, secretario de Sanidad del PSOE y portavoz de la Comisión de Sanidad del Senado.:
Kilian Sánchez, secretario de Sanidad del PSOE y portavoz de la Comisión de Sanidad del Senado.:  Rocío Hernández, consejera de Salud de Andalucía:
Rocío Hernández, consejera de Salud de Andalucía:  Nicolás González Casares, eurodiputado de Socialistas & Demócratas (S&D - PSOE):
Nicolás González Casares, eurodiputado de Socialistas & Demócratas (S&D - PSOE):  Juan José Pedreño, consejero de Salud de Murcia:
Juan José Pedreño, consejero de Salud de Murcia: 