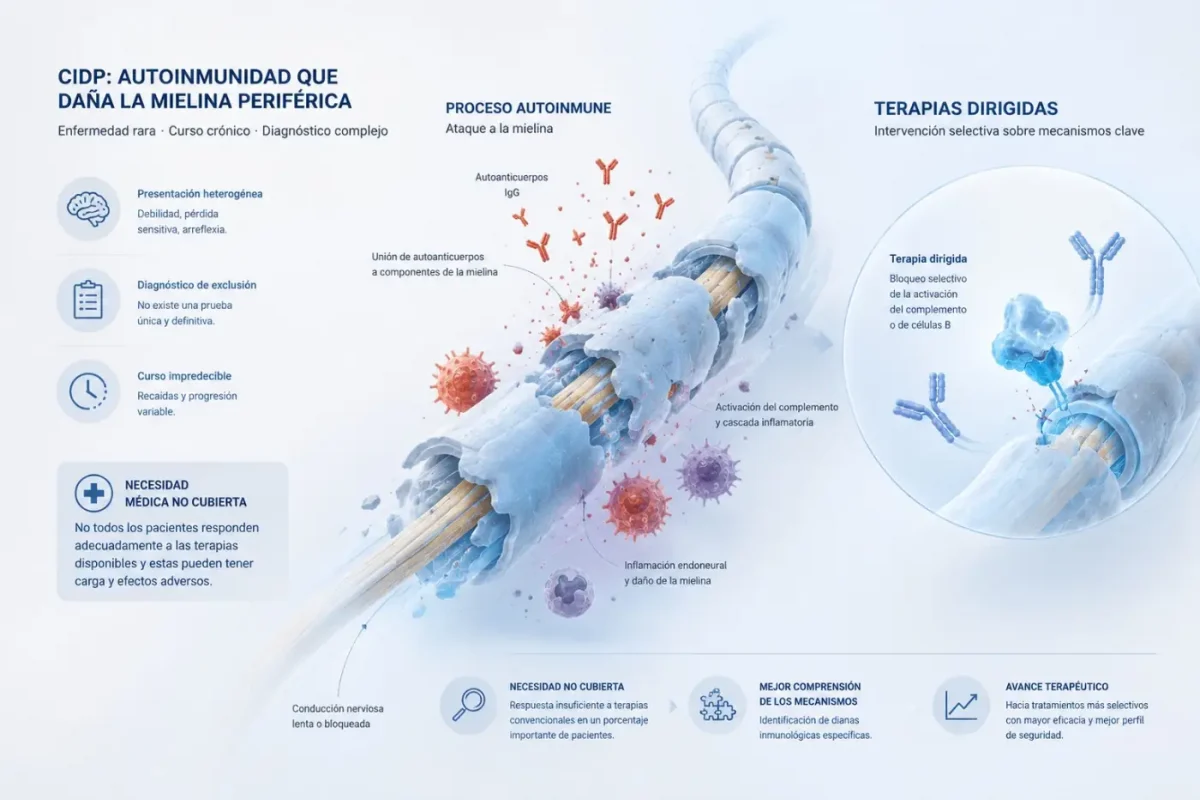
La polineuropatía desmielinizante crónica inflamatoria (CIDP, por sus siglas en inglés) es una neuropatía inflamatoria autoinmune rara del sistema nervioso periférico, caracterizada habitualmente por debilidad muscular progresiva o recurrente, afectación sensitiva y reducción o pérdida de reflejos. Los pacientes también pueden presentar dolor, fatiga y deterioro funcional relevante.
Se trata de una patología con una visibilidad limitada pese al impacto que puede tener sobre la vida de los pacientes. Sin un diagnóstico y un tratamiento adecuados, la inflamación sostenida puede favorecer daño axonal y discapacidad persistente, con afectación de la marcha, la autonomía y la calidad de vida. Un porcentaje relevante de pacientes puede evolucionar hacia una discapacidad grave que compromete la marcha y la independencia. Sin tratamiento, el 30% de los pacientes con PDIC progresan a la dependencia de silla de ruedas y, aún con el tratamiento estándar, cerca de un 13% puede llegar a necesitarla en los cinco años posteriores al diagnóstico.
El tratamiento estándar a lo largo de los últimos 30 años se ha apoyado en el uso de corticosteroides, inmunoglobulinas intravenosas (IVIg) y plasmaféresis. No obstante, una proporción relevante de pacientes no responde adecuadamente a estas terapias. Incluso estando en tratamiento, un 88% de los pacientes refieren síntomas de CIDP y secuelas, como dolor (52%) y fatiga (77%).
Esa necesidad médica no cubierta ha orientado la investigación reciente hacia terapias dirigidas que sustituyan la inmunosupresión generalizada por mecanismos más selectivos. En este sentido, la compañía inmunológica argenx es uno de los actores con cartera más avanzada en esa línea.
En la CIDP el sistema inmunológico ataca estructuras de los nervios periféricos, incluida la vaina de mielina, y en una parte de los pacientes los autoanticuerpos de tipo inmunoglobulina G (IgG) pueden desempeñar un papel relevante en ese proceso. Estos autoanticuerpos IgG actúan por dos vías: La primera es el reclutamiento de macrófagos: cuando los autoanticuerpos IgG se anclan a la mielina, los macrófagos reconocen la porción Fc de esos anticuerpos y fagocitan la mielina. La segunda es la activación del sistema del complemento: el complemento es una cascada inflamatoria que también reconoce los autoanticuerpos IgG anclados y termina atrayendo más macrófagos, que fagocitaran igualmente la mielina. Frenar ese ciclo mediante mecanismos más selectivos que la inmunosupresión generalizada es uno de los objetivos de las nuevas terapias biológicas. El correcto abordaje de la enfermedad, desde el diagnóstico a su tratamiento, requiere de una concienciación de los profesionales sanitarios acerca de la existencia de la CIPD. Actualmente no existe una prueba diagnóstica definitiva que confirme de manera absoluta la CIDP, por lo que el diagnóstico se apoya en la historia clínica, la exploración física, los estudios de conducción nerviosa y el análisis del líquido cefalorraquídeo.
Esta complejidad diagnóstica se traduce en retrasos y errores, especialmente por la similitud de sus síntomas con otras neuropatías. Una de las confusiones más relevantes se produce con el síndrome de Guillain-Barré, lo que puede demorar el inicio del tratamiento adecuado. La distinción temporal resulta clave, ya que la CIDP progresa de forma continua o recurrente durante un mínimo de ocho semanas, mientras que en el síndrome de Guillain-Barré la debilidad máxima suele alcanzarse en un periodo de una a cuatro semanas.
El retraso diagnóstico tiene consecuencias clínicas relevantes. En fases iniciales, los pacientes presentan más posibilidades de responder positivamente al tratamiento, limitar el daño en los nervios periféricos y, en algunos casos, alcanzar la remisión. Cuando la inflamación y la desmielinización se prolongan, puede producirse daño axonal. En ese escenario, la recuperación suele ser más lenta y deficiente, con riesgo de debilidad residual y pérdida permanente de función nerviosa.
El diagnóstico diferencial también condiciona la seguridad del abordaje terapéutico de la enfermedad. Es importante distinguir la CIDP típica de sus variantes y de otras enfermedades similares, como la neuropatía motora multifocal, ya que no todas responden a los mismos tratamientos. Además, en algunos casos, administrar terapias estándar de CIDP a pacientes con otras neuropatías puede resultar ineficaz o incluso empeorar la evolución clínica.
Necesidad médica persistente
Durante los últimos 30 años, el tratamiento estándar de la CIDP se apoyó en corticosteroides, inmunoglobulinas intravenosas o subcutáneas y plasmaféresis. Estas opciones pueden controlar la inflamación y mejorar la fuerza, pero no curan la enfermedad.
A pesar de las opciones terapéuticas disponibles actualmente, muchos pacientes con CIDP siguen padeciendo síntomas debilitantes, presentan fluctuaciones de la enfermedad y una carga terapéutica considerable, requiriendo a menudo tratamiento crónico. Asimismo, las opciones terapéuticas actuales pueden estar asociadas a infusiones prolongadas y complicaciones graves, como trombosis, disfunción renal aguda y otros efectos indeseables, como aumento de peso y osteoporosis, lo que limita su administración a largo plazo. Tanto las IVIg como las SCIg dependen de donantes de plasma, lo que conlleva un riesgo de escasez en la cadena de suministro.
Este conjunto de limitaciones explica que la investigación reciente se orientara hacia terapias más selectivas, capaces de actuar sobre mecanismos concretos de la enfermedad y reducir la dependencia de estrategias de inmunosupresión generalizada y que no sean dependientes de plasma.







 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


