La Agencia Europea de Medicamentos (EMA) hizo público recientemente el informe para hacer balance de los últimos 10 años en lo que respecta a la asignación de autorizaciones condicionales de comercialización, las cuales, según la Agencia, "se han convertido en una herramienta revelante para acelerar el acceso a la innovación terapéutica". No obstante, en este tiempo en el que se han concedido 36 aprobaciones condicionadas a la presentación de datos adicionales (30 entre 2006 y junio de 2016, que es lo que abarca el informe, y seis en los últimos meses de 2016: Alecensa, Lartruvo, Ninlaro, Ocaliva, Venclyxto y Zalmoxis), se ha podido comprobar también que hay margen de mejora en lo que respecta a la aceptación de esta vía por parte de los laboratorios y la solicitud, por parte de éstos, de un diálogo temprano para agilizar el proceso.
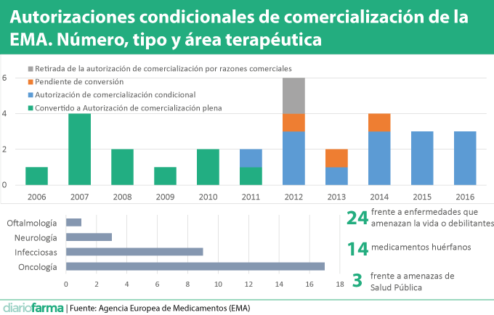
Siguiendo con las conclusiones del informe, cabe destacar que de las 30 autorizaciones condicionales concedidas (la primera fue para Sutent, el 19 de julio de 2006) en el periodo que analiza el informe, las áreas principales fueron oncología, enfermedades infecciosas, neurología y oftalmología. Cabe recordar que los requisitos para la concesión de este modelo de aprobación establecen que sólo se asignará a medicamentos para tratar, prevenir o diagnosticar enfermedades altamente debilitantes o que suponen una amenaza para la vida, a los que vienen a cubrir situaciones de emergencia o a los que se dirigen a una enfermedad rara.
De las 30 autorizaciones, sólo 14 fueron solicitadas como tales de inicio. En otros 14 casos se solicitó una vez iniciado el proceso de evaluación y 2 al reexaminar la solicitud, lo que indica, según la EMA, "la reticencia de las compañías a pedir autorización condicional de inicio". Este hecho, indican los autores del informe, ha hecho que los plazos para la concesión se retrasaran, por lo que recomiendan a los titulares de comercialización "a que busquen procedimientos de diálogo temprano" para acelerar el proceso.
El informe también distingue entre las autorizaciones condicionales que después se han convertido en autorizaciones convencionales (11). De media, esta transición suele costar unos cuatro años, según la experiencia acumulada. También están las que han sido retiradas por razones comerciales (2), y aquellas que siguen manteniendo el estatus de condicional (17), y explican que no ha habido ningún cambio de denominación entre las aprobaciones condicionadas concedidas en los últimos cinco años.
Como síntoma de la rigurosidad del procedimiento, la EMA señala que "ninguna de las 30 autorizaciones condicionales ha sido revocada o suspendida".
Evidencia que sustenta a la autorización
Además de analizar los resultados durante estos diez años, también se han presentado algunas conclusiones en lo que respeta a la evidencia que suele respaldar la concesión de una autorización condicional por parte de la EMA. Así, el informe establece que la gran mayoría de las autorizaciones contaban con resultados de estudios fase II y III abiertos, aleatorizados y con tasas de respuesta predefinidas. Los datos adicionales que se pidieron estaban casi siempre relacionados con los ensayos clínicos, los cuales, en muchos casos estaban ya en marcha y casi todos contaban con datos de eficacia y seguridad dentro de los rangos establecidos. De media, se necesitaron dos estudios abiertos, fase II, III o IV, aleatorizados para obtener una autorización condicional. "Normalmente, estos estudios requerían nuevos datos sobre el uso de tratamiento a largo plazo y/o mayor número de pacientes", añaden. Casi siempre se incluyo un fase III comparando con terapia control o placebo.
En lo que respecta a los plazos, la EMA reconoce que la dificultad para reclutar pacientes e iniciar los estudios alteró en ocasiones los plazos de autorización, aunque también se dieron algunos casos en los que se redujeron los tiempos al conseguir mejores resultados de los esperados. Concretamente, citan casos en oncología en lo relativo a la reducción de la incidencia de la metástasis o el aumento de la supervivencia.
El informe de la EMA también hace referencia a un total de 22 solicitudes de autorización condicional que no fueron aceptadas (ver expedientes en este anexo). Los motivos fueron casi siempre la percepción de un balance beneficio-riesgo negativo por carencias metodológicas que hacían poco reales los datos, insuficiencia de datos o resultados no concluyentes, con incertidumbres sobre eficacia real o falta de relevancia clínica. De ese total, 8 fueron en oncología (la que más, con mucha diferencia, después otras como reumatología y neurología con 2. El resto con 1). De las 22 solicitudes negadas, 15 se determinaron de inicio, 5 durante la evaluación y 3 al re-examinar.
Tras el balance, la EMA concluye que la autorización condicional es una vía que puede contribuir a acelerar el acceso, la cual ha ido despertando cada vez más interés entre los laboratorios, aunque no se haya reflejado en un aumento desproporcionado en el número de concesiones. En el apartado de los debe, llama a las compañías a iniciar un diálogo temprano con las autoridades para agilizar el proceso y también a "que se realicen esfuerzos adicionales en áreas en las que aún no ha tenido mucho éxito esta fórmula".







 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


