
La Agencia Española de Medicamentos y Productos Sanitarios (Aemps) ha revisado el Documento de preguntas y respuestas en relación con los dispositivos de seguridad de los medicamentos de uso humano, obligatorios desde el pasado 9 de febrero, para aclarar, entre otras cosas, cuáles son las exigencias para los fabricantes en el caso de los fármacos que se usan para la investigación clínica.
Lo ha hecho con la modificación de la pregunta 1.6, centrada, directamente, en solventar si estos requerimientos son aplicables a medicamentos destinados a ensayos. La Aemps matiza que, siempre y cuando sean productos a los que no se les ha concedido una autorización de comercialización, éstos "están excluidos de las normas relativas a los dispositivos de seguridad". Sin embargo, siempre que éstos estén autorizados tendrán que cumplir los requisitos de la Directiva 2001/83/CE y el Reglamento (UE) 2016/161 "hasta el momento en que se sepa qué lote/unidad será utilizado para los ensayos de investigación y desarrollo".
Entran en el primer grupo aquellos medicamentos fabricados "para uso conocido en un ensayo clínico", es decir, se fabrica de acuerdo con la autorización de comercialización, pero se acondiciona para un ensayo clínico. Éstos, explica la Agencia, "están excluidos de las normas relativas a los dispositivos de seguridad", aunque se requiere que el fabricante tenga una autorización de fabricación e importación (MIA) para medicamentos en investigación y que este medicamento en investigación esté certificado conforme a esa autorización de acuerdo con la solicitud del ensayo clínico, teniendo en cuenta que la solicitud del ensayo clínico debe reflejar estos acuerdos.
Otra cosa es que el medicamento esté autorizado y provenga de la cadena legal de suministro. En este caso, "deben desactivarse de conformidad con los artículos 16 y 25(4)(c) del Reglamento Delegado (UE) 2016/161 de la Comisión antes de su uso como medicamentos en investigación o como medicamentos auxiliares autorizados".
En este sentido, aclaran también, con la introducción de una nueva pregunta, la 3.7, quién debe verificar esos medicamentos, y citan los artículos 16, 23 y 25(4)(c) del Reglamento Delegado (UE) 2016/161 de la Comisión, que indica que el identificador único de medicamentos con dispositivos de seguridad en sus presentaciones comerciales debe ser verificado y desactivado "por fabricantes, mayoristas o personas autorizadas o facultadas para dispensar medicamentos al público antes de su uso como medicamentos en investigación autorizados o medicamentos auxiliares autorizados". En cuanto a los medicamentos reenvasados o reetiquetados para su uso en ensayos clínicos, será el fabricante que posee la autorización de fabricación e importación para medicamentos en investigación el que deba verificar y desactivar antes de reenvasar o reetiquetar el medicamento.
En caso de que los medicamentos no sean reenvasados o reetiquetados, será, apuntan, la persona autorizada o facultada para dispensar al público que suministra el medicamento para su uso posterior en un ensayo clínico. No obstante, recuerdan la potestad que confiere el Reglamento Delegado a los Estados miembros, "que pueden exigir a los mayoristas que verifiquen y desactiven los identificadores únicos de los medicamentos suministrados para ensayos clínicos en nombre de determinadas entidades".
En relación con los mayoristas, la Agencia también ha tratado de ayudar a éstos a saber si los medicamentos que reciben sin dispositivos de seguridad han sido liberados antes de la entrada en vigor de la normativa, el pasado 9 de febrero. La Aemps, en la respuesta 5.10, recuerda que incorporar estos dispositivos es una obligación desde esa fecha, por lo que, partiendo de la buena praxis de la industria, "eso significaría que los productos en el mercado UE sin dispositivos de seguridad deben haber sido liberados antes". No obstante, ofrecen a los mayoristas la posibilidad de "solicitar a los fabricantes que incluyan la fecha de liberación del lote en el albarán de entrega de medicamentos no serializados, para confirmarlo".




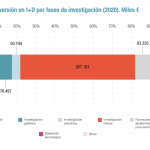












 :
:  Carmen Martínez, portavoz del Grupo Socialista en la Comisión de Sanidad del Congreso de los Diputados:
Carmen Martínez, portavoz del Grupo Socialista en la Comisión de Sanidad del Congreso de los Diputados:  Margarita de la Pisa, eurodiputada de Vox.:
Margarita de la Pisa, eurodiputada de Vox.:  Ion Arocena, director general de la Asociación Española de Bioempresas (Asebio):
Ion Arocena, director general de la Asociación Española de Bioempresas (Asebio):  Javier Padilla, secretario de Estado de Sanidad:
Javier Padilla, secretario de Estado de Sanidad: