La diferencia entre las autorizaciones de medicamentos innovadores por parte de la FDA estadounidense y la Agencia Europea de Medicamentos (EMA) se ha ampliado de forma continua a lo largo de los últimos años. Así se desprende del último informe Regulatory Approval Indicator, elaborado por IQVIA para EFPIA, que analiza la evolución de las nuevas sustancias activas aprobadas por la FDA y su posterior autorización por parte de otros reguladores internacionales.
El dato central del informe es especialmente relevante desde el punto de vista de política farmacéutica ya que la proporción de nuevas sustancias activas aprobadas por la FDA que también contaban con aprobación de la EMA ha pasado del 77% en la cohorte de 2021 al 38% en la cohorte de 2025. Esta situación no parece que vaya a revertirse sino que, por el contrario, podría incrementarse debido a las políticas proteccionistas de Donald Trump en la Casa Blanca y, especialmente su ‘Nación Más Favorecida’ (MFN).
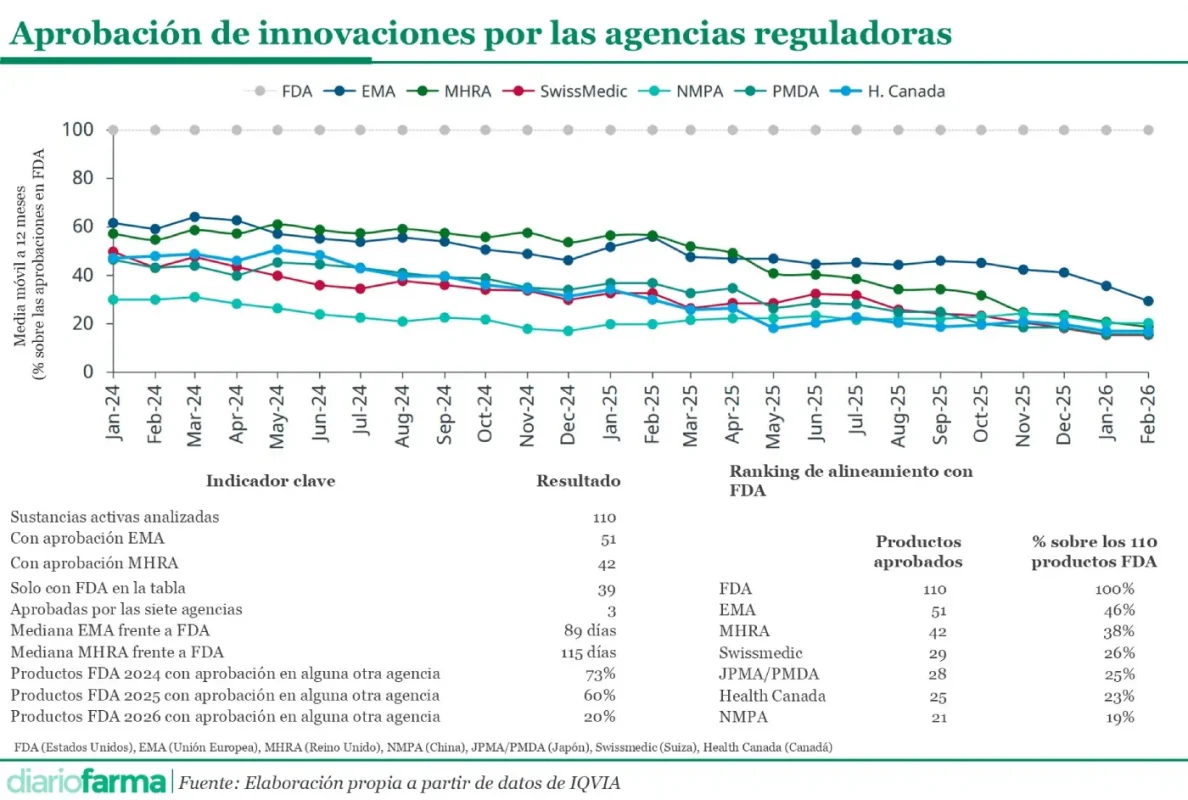
El informe de IQVIA analiza un total de 258 nuevas sustancias activas (NAS) aprobadas por la FDA entre 2021 y febrero de 2026. Sobre ese universo, la consultora rastrea si esos mismos productos han obtenido autorización en seis jurisdicciones: EMA (Unión Europea), MHRA (Reino Unido),NMPA (China), PMDA (Japón), Swissmedic (Suiza), Health Canada (Canadá).
Según el documento, con datos actualizados a abril de 2026, la EMA acumula 159 aprobaciones sobre ese conjunto de productos, por delante de MHRA (145), Swissmedic (110), PMDA (107), Health Canada (98) y NMPA (73) de ese total de 258 aprobaciones en Estados Unidos.
La lectura que plantea el documento es que Europa sigue siendo uno de los principales mercados regulatorios de referencia, pero su grado de coincidencia con las aprobaciones estadounidenses se ha reducido de forma notable. El informe apunta, además, a que el deterioro de la cuota europea se acentúa en la segunda mitad de 2025, según la media móvil de 12 meses incluida en el documento, algo que coincide con la implantación de la MFN.
El informe, que se publicó conjuntamente con el Estudio WAIT la semana pasada, no mide disponibilidad efectiva para los pacientes, ni tiempos de financiación, ni precio, ni reembolso. Su foco está exclusivamente en el plano regulatorio y compara si los medicamentos aprobados por la FDA han recibido también autorización por parte de otros reguladores.
Este matiz es relevante para interpretar correctamente los datos. La autorización de la EMA constituye un hito regulatorio europeo, pero el informe no analiza las fases posteriores de acceso en los Estados miembros.
Asimetría en las cohortes recientes
Más allá de la lectura agregada, el informe incluye un anexo que permite observar producto a producto qué ocurre con las nuevas sustancias activas aprobadas por la FDA en 2024, 2025 y enero-febrero de 2026 y sus fechas de aprobación en cada agencia.
Una análisis de ese anexo muestra que, sobre 110 medicamentos o nuevas sustancias activas incluidos en esas cohortes recientes, 59 aparecen con aprobación de la FDA pero sin fecha de autorización EMA. Esto equivale aproximadamente al 54% del total. Los 51 restantes cuentan con fecha tanto de la FDA como de la EMA.
Entre esos 51 productos aprobados por ambos reguladores, la FDA fue antes en 37 casos, mientras que la EMA se adelantó en 14. Esta lectura refuerza la idea de una ventaja temporal frecuente para Estados Unidos, aunque también introduce un matiz relevante: Europa no siempre llega después.
La evolución por cohortes dentro del anexo apunta en la misma dirección que el indicador general. En la cohorte FDA de 2024, 23 de los 55 productos incluidos no han sido aprobados por la EMA, lo que representa un 42%. En la cohorte de 2025, la cifra asciende a 32 de 50, un 64%. En enero-febrero de 2026, 4 de los 5 productos incluidos en la FDA no cuentan tienen reflejo en Europa, aunque este último dato debe interpretarse con especial cautela por tratarse de una muestra muy reducida y de productos muy recientes.
Desfases entre las autorizaciones de FDA y EMA
El detalle del anexo permite identificar varios medicamentos en los que la autorización estadounidense precede en más de un año a la europea.
Entre los mayores desfases figuran nogapendekin alfa inbakicept, aprobado por la FDA el 22 de abril de 2024 y por la EMA el 16 de febrero de 2026, con una diferencia de 665 días; resmetirom, con aprobación FDA el 14 de marzo de 2024 y EMA el 18 de agosto de 2025, con 522 días de diferencia; y levacetylleucine, aprobado por la FDA el 24 de septiembre de 2024 y por la EMA el 19 de enero de 2026, con 482 días.
También destacan donanemab, con una diferencia de 449 días entre FDA y EMA; givinostat, con 442 días; y vorasidenib, con 407 días.
Estos casos ilustran la dimensión práctica de la diferencia temporal entre las autorizaciones de ambas agencias cuando la aprobación europea llega después de la estadounidense.
No obstante, el informe no permite atribuir automáticamente estos desfases a una única causa. El documento recoge fechas de aprobación, pero no incorpora para cada producto la fecha de presentación del expediente ante la EMA, la duración neta de evaluación, posibles paradas de reloj, diferencias en la indicación solicitada o decisiones estratégicas de las compañías.
Europa también se adelanta en algunos productos
El anexo también muestra casos en sentido contrario. Hay 14 productos en los que la aprobación de la EMA aparece antes que la de la FDA.
El ejemplo más acusado es treosulfan, con autorización EMA el 20 de junio de 2019 y aprobación FDA el 21 de enero de 2025, una diferencia de 2.041 días. También destacan atidarsagene autotemcel, autorizado por la EMA el 17 de diciembre de 2020 y por la FDA el 18 de marzo de 2024, con 1.187 días de diferencia; eladocagene exuparvovec, con 849 días; y pegzilarginase-nbln, con 801 días.
Otros casos en los que la EMA se adelanta son vadadustat, delgocitinib, lebrikizumab, palopegteriparatide, tislelizumab, garadacimab, linvoseltamab, sepiapterin, zolbetuximab y concizumab.
Huérfanos y oncología pesan en la composición de las cohortes
El propio informe advierte de que la composición de las cohortes puede influir en la comparación entre reguladores. En los años analizados, los medicamentos huérfanos y los productos oncológicos representan una parte relevante de las aprobaciones de la FDA.
En 2021, de las 50 aprobaciones FDA incluidas en el indicador, 29 correspondían a productos huérfanos, un 58%, y 16 a oncología, un 32%. En 2022, los huérfanos fueron 21 de 35, un 60%, y oncología 13, un 37%. En 2023, se registraron 35 huérfanos sobre 63 aprobaciones, un 55%, y 18 oncológicos, un 29%.
La pauta se mantiene en 2024, con 33 huérfanos sobre 55 aprobaciones FDA, un 60%, y 17 productos oncológicos, un 31%. En 2025, el peso de los huérfanos baja al 40%, con 20 productos sobre 50, mientras que oncología representa el 28%, con 14 productos.
Esta composición es relevante porque los medicamentos huérfanos, oncológicos o dirigidos a nichos terapéuticos pueden seguir calendarios regulatorios y estrategias de autorización diferentes. El informe señala que las diferencias en las cuotas de aprobación entre países pueden estar condicionadas por el peso de este tipo de productos.








 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


