El Real Decreto por el que se regula la Evaluación de las Tecnologías Sanitarias (ETS), aprobado por el Consejo de Ministros y que ha sido publicado este viernes en el Boletín Oficial del Estado (BOE), llega con un balance favorable a la seguridad jurídica del procedimiento respecto al borrador sometido a audiencia pública. Las modificaciones introducidas en la recta final, después del dictamen del Consejo de Estado, refuerzan las garantías de los desarrolladores en dos planos. Por un lado, el reconocimiento de un trámite de audiencia con cobertura legal plena y la elevación a rango de orden ministerial del grueso de las instrucciones que regirán el sistema. Como contrapartida, los ministerios económicos del Estado entran con peso propio en los órganos del nuevo modelo, en un movimiento que reequilibra su gobernanza interna.
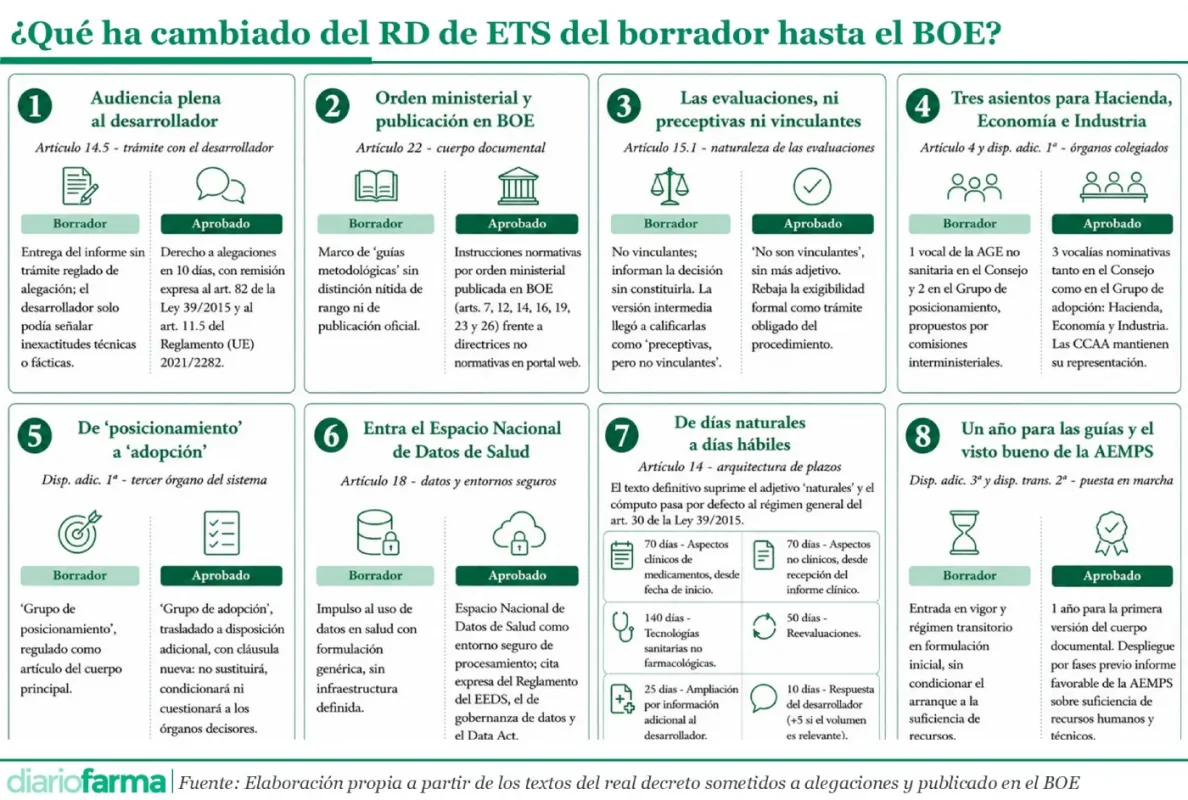
El cambio de mayor calado jurídico para el sector se encuentra en el artículo 14.5. Los textos iniciales, incluso el que se remitió al Consejo de Estado, limitaban la capacidad del desarrollador a señalar “inexactitud meramente técnica o fáctica” del proyecto de informe y excluía expresamente que pudiera formular observaciones sobre los resultados de la evaluación. El texto definitivamente aprobado sustituye ese marco restrictivo por un derecho pleno a presentar alegaciones en un plazo máximo de diez días, con remisión expresa al artículo 82 de la Ley 39/2015 de Procedimiento Administrativo Común y al artículo 11.5 del Reglamento (UE) 2021/2282. El salto es cualitativo ya que el papel del desarrollador pasa de corregir erratas a un trámite de audiencia procedimental ordinario en el sentido del derecho administrativo común.
Esta apertura del procedimiento al desarrollador fue una de las demandas centrales de Farmaindustria en el trámite de alegaciones, una petición que el Ministerio de Sanidad no recogió en el texto que elevó al Consejo de Estado y que sólo cristalizó en la versión final, tras el dictamen del alto órgano consultivo.
Órdenes ministeriales para las guías
A la mejora del trámite de audiencia se suma la reordenación del cuerpo documental y las guías que vertebrarán todo el sistema. El artículo 22 del texto aprobado distingue con nitidez entre instrucciones normativas, que se aprobarán mediante orden ministerial de la persona titular de Sanidad, como publicó Diariofarma, y se publicarán en el Boletín Oficial del Estado, y directrices metodológicas de carácter no normativo, aprobadas por el Consejo de ETS y publicadas en el portal web del Ministerio. Las primeras desarrollarán el contenido sustantivo de varios artículos, en concreto, 7, 12, 14, 16, 19, 23 y 26, que regulan modelos de expediente, reglas de presentación electrónica, plazos de subsanación, procedimiento de publicación, consultas científicas, obligaciones del desarrollador y régimen de conflicto de interés. La distinción no es menor ya que lo que crea obligaciones para terceros exige ahora rango reglamentario y publicación oficial, una mejora que responde a la lógica de las observaciones que suelen acompañar a este tipo de dictámenes. Esta fue otra de las demandas más relevantes de Farmaindustria, así como de numerosos juristas expertos en la materia.
Otro de los cambios de relevancia se debe a la mención explícita en el artículo 15.1 de que las evaluaciones “no serán vinculantes” ni serán preceptivas. Aquí se ha producido un nuevo cambio respecto de la redacción que se envió al Consejo de Estado donde se introdujo que los informes “son preceptivos, pero no vinculantes”, incorporando esa exigencia respecto del texto inicial sometido a alegaciones. Aunque el matiz parezca menor, rebaja la exigibilidad formal de la evaluación como trámite obligado dentro del procedimiento de financiación y precio, y reduce el riesgo de que su ausencia se convierta en obstáculo formal para la decisión.
Pese a esta eliminación, podría entenderse que el artículo 6 ya establece qué tipos de tecnologías serán objeto de evaluación, si bien la eliminación de esa acotación del artículo 15.1 parece rebajar la necesidad de evaluación. Eso sí, los desarrolladores están obligados a presentar toda la documentación exigida si quieren tramitar ser financiados en España. Además, cabe señalar una discrepancia con lo que el Ministerio de Sanidad señaló en su nota de prensa tras el Consejo de Ministros, donde se indicó que las evaluaciones “tendrán carácter preceptivo, pero no vinculante”, como se recogía en la versión que entró en el Consejo de Estado, pero no en la publicada en el BOE. No es la única contradicción.
En cuanto al alcance de la evaluación, cabe señalar otra novedad en los dominios no clínicos que se tendrán que analizar. El borrador sometido a alegaciones establecía no recogía el detalle de los mismos, si bien el texto remitido al Consejo de Estado detallaba que serían aspectos éticos, organizativos, sociales, jurídicos y ambientales. En el texto final, a todos estos se añaden los de género, que tendrán que ser evaluados igualmente.
Tres asientos para los ministerios económicos
El contrapeso a las mejoras de garantías llega por la vía de la composición de los órganos colegiados. El texto original preveía un único representante de la Administración General del Estado ajeno a Sanidad en el Consejo de Gobernanza, designado a propuesta conjunta de la Comisión Interministerial de Precios de los Medicamentos (CIPM) y la de Prestaciones, algo que se conservó en el texto remitido al Consejo de Estado. No obstante, la versión aprobada eleva esa presencia a tres vocalías nominativas, designadas respectivamente por los ministerios de Hacienda, de Economía, Comercio y Empresa, y de Industria y Turismo.
El mismo movimiento se reproduce en el Grupo de adopción, donde las dos vocalías, a repartir entre los ministerios presentes en la CIPM distintos a sanidad, que figuraban en diciembre se convierten en tres asientos propios para los tres departamentos económicos, tanto en la configuración de medicamentos como en la de tecnologías no farmacológicas. La representación autonómica, en cambio, se mantiene inalterada respecto al borrador inicial, con seis vocalías en el Consejo de ETS y una por comunidad autónoma en el Grupo de adopción.
De posicionamiento a adopción
Ya era conocido el cambio de denominación del tercer órgano del sistema, que el borrador llamaba ‘Grupo de posicionamiento’ y que en el texto aprobado pasa a ser ‘Grupo de adopción’. Lo nuevo del texto definitivo es la cláusula que cierra su régimen. La disposición adicional primera precisa ahora que el grupo “en ningún caso sustituirá, condicionará ni cuestionará las competencias de los órganos decisores”. El blindaje refuerza la idea, repetida en todo el articulado, de que el sistema de ETS informa pero no decide, y reduce el margen para interpretaciones expansivas de ese órgano colegiado.
La financiación del sistema es otra de las novedades. Regulada en el artículo 27, también muta entre el borrador inicial y el texto final en una dirección que conecta con el papel reservado a la Aemps. La versión sometida a audiencia pública comprometía una financiación “suficiente, justa y ajustada al trabajo desarrollado”. El texto aprobado elimina esa declaración y la sustituye por una fórmula posibilista en el que la financiación “podrá articularse” a través de los instrumentos previstos en el ordenamiento, entre ellos el sistema de aportaciones por volumen de ventas al SNS, los Presupuestos Generales del Estado o tasas y precios públicos, todo ello “ajustado a las disponibilidades presupuestarias existentes”.
El desplazamiento del compromiso de suficiencia a la sujeción al crédito disponible explica, en buena medida, que el despliegue por fases quede condicionado al informe favorable previo de la Agencia Española de Medicamentos y Productos Sanitarios (Aemps) sobre los recursos humanos y técnicos disponibles. En este sentido, la disposición transitoria segunda condiciona el despliegue por fases a una resolución de la Dirección General de Cartera Común de Servicios del SNS y Farmacia que requerirá informe favorable previo de la Aemps.
La Agencia, además de representar a España en el Grupo de Coordinación europeo sobre ETS y de albergar la Oficina de Evaluación de Medicamentos como unidad funcional propia, queda así situada en un papel doble de ejecutor del nuevo modelo y validador de su viabilidad operativa antes del arranque.
Un año de transitoriedad y plazos
La aplicación efectiva del modelo no será inmediata. La disposición adicional tercera fija el plazo de un año desde la entrada en vigor. Este plazo será esencial para aprobar la primera versión de las instrucciones normativas y publicar las directrices metodológicas.
El artículo 14 mantiene en el texto definitivo la arquitectura de plazos que ya figuraba en versiones intermedias, aunque los contabiliza como hábiles en vez de naturales, como aparecía en la versión sometida a alegaciones. Al suprimir el adjetivo “naturales” del articulado, el cómputo pasa por defecto al régimen general de plazos administrativos previsto en la Ley 39/2015. De este modo, los plazos quedan fijados del siguiente modo: aspectos clínicos de medicamentos, 70 días desde la fecha de inicio, sin rebasar los 15 días desde la publicación del informe conjunto europeo cuando proceda; aspectos no clínicos de medicamentos, 70 días desde la recepción del informe sobre los aspectos clínicos, con posibilidad de solapar ambas evaluaciones cuando la disponibilidad parcial lo permita; reevaluaciones, 50 días; ampliación por información adicional, 25 días, cuando las oficinas necesiten recabar especificaciones, aclaraciones o datos adicionales. Por su parte, las alegaciones de la industria tendrán unos plazos de 10 días, prorrogables en cinco más si el volumen de trabajo resulta relevante.
Aquí es donde se produce otra discrepancia con respecto a la nota de prensa del Ministerio, que hablaba de días naturales para el cómputo de los plazos: “el sistema establece un plazo máximo de 90 días naturales para la elaboración de los informes sobre aspectos clínicos y de otros 90 días naturales para los aspectos no clínicos en el caso de los medicamentos, mientras que las tecnologías sanitarias no farmacológicas contarán con un plazo máximo de evaluación de 180 días”.
Conflictos de interés
Por otro lado, el régimen de conflictos de interés del artículo 26 también gana cuerpo en el texto final. La norma define ahora con detalle que se considera conflicto la participación en actividades de asesoría científica, estratégica o técnica realizada para la industria de las entidades desarrolladoras de forma directa o indirecta, y la pertenencia a órganos de dirección, asesoramiento o financiación vinculados a dichas entidades. Se excluyen los trabajos de consultoría que previamente estaban listados como causa de conflicto de interés. Una orden ministerial específica publicará los documentos de declaración, sus efectos sobre la participación en las evaluaciones y los periodos de incompatibilidad aplicables.
Entre las novedades sustantivas del texto definitivo figura también la mención expresa al Espacio Nacional de Datos de Salud como entorno de procesamiento seguro para la evaluación, que el artículo 18 incorpora junto a la cita del Reglamento del Espacio Europeo de Datos de Salud, el Reglamento de gobernanza de datos y el Data Act. La concreción de la infraestructura, ausente con esa claridad en la versión presentada al Consejo de Estado, da soporte normativo al uso secundario de datos en las evaluaciones, una pieza considerada crítica para alinear el modelo español con el Reglamento europeo de ETS.







 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


