
INTRODUCCIÓN
Un medicamento elaborado industrialmente necesita la autorización previa de la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) o de la Agencia Europea del Medicamento (EMA), en el caso de que se haya seguido un procedimiento centralizado de autorización, para poder comercializarse. En cualquiera de estos casos, el medicamento habrá tenido que demostrar ser seguro y eficaz durante la fase de ensayos clínicos. No obstante, la información sobre seguridad obtenida durante esta etapa no es suficiente y es necesario seguir recopilando datos una vez que el medicamento ya está en el mercado porque será entonces cuando se esté utilizando en las condiciones clínicas habituales, por un número de personas mucho más amplio, en grupos de población concretos (ancianos, niños, etc.), durante un periodo de tiempo mayor, etc.
Según la Organización Mundial de la Salud (OMS) la farmacovigilancia “es la ciencia y actividades relacionadas con la detección, evaluación, comprensión y prevención de efectos adversos o cualquier otro problema relacionado con los medicamentos”.
Esta definición no sólo se refiere a lo que, a priori, podría ser el objetivo principal de esa disciplina según la legislación española, los efectos o reacciones adversas a medicamentos1, sino que amplía su radio de acción para incluir otros aspectos relacionados con la seguridad y la efectividad de los medicamentos como pueden ser medicamentos de baja calidad, medicamentos falsificados, errores de medicación, falta de efectividad, uso en indicaciones no recogidas en ficha técnica, mal uso, abuso de medicamentos, interacciones, efectos a largo plazo, efectos latentes, impacto medioambiental del uso del medicamento, efectos sobre la salud humana de residuos de medicamentos en animales, etc.
Igualmente, en los últimos años se han empezado a realizar tareas de farmacovigilancia con medicamentos de origen biológico, derivados de sangre, vacunas, medicamentos a base de plantas, medicamentos tradicionales, tratamientos alternativos o productos sanitarios. La vigilancia de medicamentos nuevos o de medicamentos que requieran de seguimiento adicional será más exhaustiva
Esta evolución de la farmacovigilancia ha necesitado del desarrollo de unos sistemas nacionales de farmacovigilancia efectivos y coordinados y de una implicación cada vez más elevada de todos los profesionales de la salud.
1Reacción adversa es cualquier respuesta nociva y no intencionada a un medicamento. RD 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano.
El Sistema Español de Farmacovigilancia de medicamentos de uso humano (SEFV–H) es una estructura descentralizada, formada por los organismos autonómicos competentes en farmacovigilancia y la AEMPS y en el que participan la industria farmacéutica, los profesionales sanitarios y los propios pacientes a través de la remisión de notificaciones de sospechas de reacciones adversas a medicamentos de uso humano, catalogándose la información en la base de datos FEDRA. Además, España forma parte del programa de farmacovigilancia de la UE (EudraVigilance2) y también de la OMS (VigiBase3) donde se recibe información de 118 centros nacionales que es analizada y posteriormente distribuida de nuevo a las agencias de medicamentos nacionales.

En base a la información recibida, la AEMPS (en colaboración con la EMA si fuera necesario), podrá determinar si es necesario modificar las condiciones de autorización de un medicamento y reflejarlo consecuentemente en su prospecto y ficha técnica, suspenderlo o revocar dicha autorización de comercialización.
La principal fuente de información sobre sospechas de reacciones adversas son los profesionales sanitarios, si bien desde julio de 2012 los pacientes también pueden participar en el sistema de notificación espontánea de sospechas de reacciones adversas (aunque siguen un camino distinto al de los profesionales). La incorporación de los pacientes puede servir para ampliar el conocimiento y la percepción sobre problemas de seguridad relacionados con los medicamentos. La notificación por parte del propio paciente puede realizarse directamente a través del formulario de notificación electrónico, disponible en www.notificaRAM.es o pueden seguir haciendo la notificación a través de un profesional sanitario.
2 https://eudravigilance.ema.europa.eu/human/index.asp
Por otro lado, la circulación de medicamentos falsificados4 es un problema cada vez mayor. La OMS estima que en países desarrollados, con buenos sistemas de detección, menos del 1% de los medicamentos disponibles son medicamentos falsificados, si bien esta cifra aumenta de forma significativa y es variable en países en vías de desarrollo. Aunque en España la cadena de distribución es muy segura, el farmacéutico debe estar alerta ante la aparición de posibles medicamentos falsificados, en especial por la venta a distancia a través de sitios web.
El farmacéutico comunitario ocupa una posición idónea para ejercer actividades de farmacovigilancia y de detección de medicamentos falsificados.
El presente documento recoge las recomendaciones necesarias para que la práctica de las actividades relacionadas con la farmacovigilancia y la prevención de la entrada de medicamentos falsificados a la cadena legal de suministro por parte del farmacéutico comunitario puedan considerarse buenas prácticas profesionales.
4Medicamento falsificado es cualquier medicamento cuya presentación sea falsa respecto a su identidad, origen o historial y no comprende ni defectos de calidad involuntarios y se entiende sin perjuicio de las violaciones de los derechos de propiedad intelectual. Directiva 2011/62/UE.
OBJETIVOS DE LAS ACTIVIDADES DIRIGIDAS A LA FARMACOVIGILANCIA Y LA PREVENCIÓN DE ENTRADA DE MEDICAMENTOS FALSIFICADOS EN LA CADENA LEGAL DE SUMINISTRO
El objetivo general de este servicio es promover el uso seguro, efectivo y responsable de los medicamentos.
Los objetivos concretos son:
- a) Detectar e informar a los servicios de Farmacovigilancia autonómicos y/o nacionales sobre la seguridad, las sospechas de reacciones adversas relacionadas con el uso de medicamentos y la Salud Pública incluyendo aspectos de calidad, medicamentos falsificados, errores de medicación, mal uso/abuso de medicamentos, efectos a largo plazo, efectos latentes, impacto medioambiental del uso de medicamentos o efectos sobre la salud humana de residuos de medicamentos veterinarios.
- b) Prevenir el daño de las reacciones adversas asociadas al uso de medicamentos, tanto bajo las condiciones de uso aprobadas, como bajo otras condiciones.
- I. PROCEDIMIENTO DE NOTIFICACIÓN DE SOSPECHAS DE REACCIONES ADVERSAS A MEDICAMENTOS POR PARTE DEL FARMACÉUTICO COMUNITARIO
Ante la sospecha de una posible reacción adversa a un medicamento5, el farmacéutico debe:
- 1. Conocer qué debe notificarse al SEFV–H: se notificará cualquier sospecha de reacción adversa producida por cualquier tipo de medicamento (incluyendo vacunas, medicamentos de origen biológico, radiofármacos, medicamentos publicitarios, medicamentos homeopáticos, medicamentos a base de plantas y fórmulas magistrales) si bien se dará prioridad a sospechas de reacciones adversas a medicamentos sujetos a seguimiento adicional, sospechas de reacciones adversas graves o inesperadas y sospechas de reacciones adversas en niños con independencia de si el medicamento está aprobado o no para su uso en población pediátrica, embarazadas y ancianos.
Se notificará cualquier sospecha, independientemente de si el medicamento se ha utilizado según las indicaciones descritas en ficha técnica o no.
5 En este procedimiento se aplica la acepción amplia de reacción adversa, incluyéndose todos aquellos aspectos susceptibles de notificación reflejados en la introducción de este documento.
Los medicamentos sujetos a seguimiento adicional estarán identificados con el símbolo (▼) común para todo el ámbito de la UE y que aparecerá en el prospecto y en la ficha técnica del medicamento. Los medicamentos sujetos a seguimiento adicional son:
₋ Todos los medicamentos autorizados después del 1 de enero de 2011 que contengan un nuevo principio activo.
₋ Medicamentos de origen biológico como vacunas o productos derivados de plasma, autorizados después del 1 de enero de 2011.
₋ Productos para los que se necesite información adicional post-autorización o para los cuales la autorización esté sujeta a condiciones o restricciones basadas en su seguridad y uso efectivo.
₋ Medicamentos que, a criterio del Comité para la Evaluación de Riesgos en Farmacovigilancia (PRAC) de la Agencia Europea del Medicamento (EMA), deban someterse a seguimiento adicional.
Estos medicamentos permanecerán en seguimiento adicional durante al menos 5 años o el tiempo que el PRAC considere necesario. El listado de estos medicamentos está disponible en la página web de la EMA y es revisado mensualmente por el PRAC.
Las reacciones adversas graves son cualquier reacción adversa que ocasione la muerte, pueda poner en peligro la vida, exija la hospitalización del paciente o la prolongación de una hospitalización ya existente, ocasione una discapacidad o invalidez significativa o persistente o constituya una anomalía congénita o defecto de nacimiento. A efectos de notificación, se tratarán también como graves aquellas sospechas de reacción adversa que se consideren importantes desde un punto de vista médico, aunque no cumplan con los criterios anteriores.
Las reacciones adversas inesperadas son cualquier reacción adversa cuya naturaleza, gravedad o consecuencias no sean coherentes con la información descrita en la ficha técnica del medicamento.
Respecto a la notificación de sospechas de reacciones adversas en población pediátrica y mujeres embarazadas, se recomienda en todo momento dado que de forma habitual no suelen realizarse ensayos clínicos con este tipo de poblaciones por lo que se dispone de menos información sobre posibles reacciones adversas a medicamentos. En el caso de las personas mayores pueden ser más susceptibles dado que metabolizan o excretan los medicamentos con menos eficacia y pueden ser más sensibles a sus efectos.
En ocasiones puede ser difícil conocer la relación de causalidad entre la aparición de una reacción adversa y el uso de un medicamento. A la hora de evaluar esta causalidad se pueden tener en cuenta factores como la relación temporal, la naturaleza de la reacción, el efecto de la re-exposición al medicamento, la relación dosis-respuesta, el efecto de la interrupción del uso del medicamento sospechoso, etc.
A pesar de esto, se recomienda notificar cualquier tipo de sospecha de reacción adversa o evento susceptible de ser notificado.
En el caso de aspectos en el etiquetado que puedan provocar errores de medicación, deberá recogerse todo aquello que pudiera inducir a dicho error, por ejemplo:
₋ Marcas de medicamentos que generen confusión por similitud (medicamentos parónimos).
₋ Nombres de medicamentos que sean difíciles de distinguir, bien por la dosis o por la vía de administración.
₋ Etiquetado que incluya texto confuso o poco visible para el usuario.
₋ Información confusa o limitada para el paciente (prospecto) o para el profesional sanitario (ficha técnica).
En caso de que se quiera hacer partícipe a la comunidad científica mediante publicación, ésta habrá de hacerse según guías consensuadas para publicar reacciones adversas a fármacos.
- Cumplimentar el formulario de notificación de sospecha de reacción adversa: actualmente existen dos vías para realizar la notificación, o bien utilizando la denominada tarjeta amarilla (formato papel) o bien a través del formulario de notificación electrónico disponible en www.notificaRAM.es. Sin perjuicio de que en el futuro se puedan utilizar otros sistemas de notificación, como por ejemplo, el sistema de receta electrónica.
La información que es necesario incluir en la notificación se puede dividir en 4 secciones principales:
- Información del paciente:
₋ Nombre y apellidos, al menos el primer apellido, o iniciales.
₋ Número de tarjeta sanitaria o de la historia clínica para ayudar a la identificación del paciente en caso de duplicaciones o de notificaciones adicionales (recomendable).
₋ Sexo.
₋ Edad (incluyendo horas, días, semanas, meses, etc. en el caso de bebés) o grupo de edad (recién nacido, lactante, niño, adolescente, adulto, anciano).
₋ Peso (kg) y altura (cm) aproximados.
La información personal no infringe acuerdos de confidencialidad entre el paciente y el farmacéutico ya que la Ley 29/2006 de garantías y uso racional de medicamentos obliga a los profesionales sanitarios a notificar cualquier sospecha de reacción adversa a un medicamento.
- b. Medicamento sospechoso:
₋ Nombre comercial del medicamento o principio activo para el caso de medicamentos genéricos. Para medicamentos homeopáticos, a base de plantas o fórmulas magistrales, indicar la composición en la forma más detallada posible.
₋ Dosis, forma farmacéutica, presentación y marca o laboratorio fabricante.
₋ Número de lote y fecha de caducidad.
₋ Motivo de la prescripción, es decir, la indicación del medicamento si se conoce.
₋ Dosis diaria y pauta posológica. En el caso de vacunas que se administren en varias dosis, indicar en cuál de dichas dosis se ha producido la sospecha de reacción adversa.
₋ Vía de administración.
₋ Medidas tomadas, es decir, si se ha retirado el medicamento, disminuido la dosis, aumentado la dosis, etc.
₋ Fechas de inicio/fin de tratamiento.
- Reacción adversa identificada:
₋ Gravedad: es decir si la reacción adversa ha puesto en peligro la vida del paciente, ha sido causa de hospitalización o prolongación de una hospitalización, ha provocado una incapacidad, anomalía congénita o defecto grave o la muerte del paciente.
₋ Descripción de la reacción adversa incluyendo signos y síntomas.
₋ Tipo de notificación: si es espontánea o si es una sospecha de reacción adversa producida durante la realización de un estudio.
₋ Si la reacción identificada está relacionada con un error de medicación6 se indicará y describirá en este apartado.
₋ Tratamiento empleado, si aplica.
₋ Fecha de inicio/fin.
₋ Desenlace de la reacción: recuperado/resuelto, en recuperación/resolución, no recuperado/no resuelto, recuperado/resuelto con secuelas, no recuperado/resuelto, muerte o se desconoce.
6 Error de medicación es un fallo no intencionado en el proceso de prescripción, dispensación o administración de un medicamento bajo el control del profesional sanitario o del ciudadano que consume el medicamento. Los errores de medicación que ocasionen un daño al paciente se consideran reacciones adversas, excepto aquellos derivados del fallo terapéutico por omisión de un tratamiento (RD 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano).
- d. Información del notificador:
₋ Nombre y apellidos.
₋ Profesión.
₋ Centro de trabajo.
₋ Dirección del centro de trabajo (población, provincia, código postal).
₋ Teléfono de contacto.
₋ Correo electrónico.
- e. Información adicional: se incluirá cualquier información que se considere relevante para facilitar la comprensión y la evaluación de la sospecha de reacción advers
₋ Medicamentos empleados en los meses previos a la aparición de la reacción adversa o que se estén empleando en el momento de su aparición.
₋ Información sobre re-exposición al medicamento sospechoso, si existiese.
₋ Antecedentes o datos que pudieran ser de interés, alergias, parámetros biológicos relevantes, etc.
₋ En el caso de anomalías congénitas, incluir toda la medicación que se haya podido utilizar durante el embarazo indicando fechas aproximadas o trimestre. También indicar la fecha aproximada en la que se produjo la última menstruación.
₋ Alimentos ingeridos que se sospeche puedan interaccionar con el medicamento.
₋ Información sobre uso no autorizado, sobredosis, mal uso del medicamento, abuso, uso de medicamentos no autorizados, medicamentos extranjeros, etc.
Es recomendable realizar una copia de la notificación remitida y conservarla durante algún tiempo para poder completar o realizar seguimiento en caso necesario.
- 3. Enviar la notificación de sospecha de reacción adversa: en función de la vía de notificación elegida el envío se realizará bien por correo postal al centro autonómico de farmacovigilancia correspondiente o, en el caso de utilizar el formulario electrónico, se indicará la comunidad autónoma desde donde se manda la notificación y el sistema informático redireccionará al centro autonómico las sospechas enviadas.
La notificación de la sospecha de reacción adversa se enviará tan pronto como sea posible.
El centro autonómico de farmacovigilancia remitirá un informe de acuse de recibo en el que se incluirá un número de referencia. Este número de referencia deberá indicarse en el caso de modificaciones o ampliaciones de información ya enviada.
En el centro se analiza la información para determinar si hay nuevos riesgos o un cambio en la gravedad o frecuencia de riesgos ya conocidos.
Cada centro autonómico está conectado a FEDRA, la base de datos del SEFV-H en la que se alojan todos los casos de sospechas de reacciones adversas a medicamentos.
Luego FEDRA transmite a las bases internacionales. EudraVigilance (Módulo EVPM para notificaciones postautorización) y VigiBase (Centro de Monitorización de Uppsala de la OMS).
En el caso de notificaciones relacionadas con aspectos del etiquetado y/o material de acondicionamiento de un medicamento, se incluirán todos aquellos aspectos que puedan conducir a errores de medicación y contener la mayor cantidad de información posible para permitir una evaluación del acaso y la pertinente toma de decisiones. La notificación puede ser remitida por cualquier persona interesada, paciente, profesional, etc.
La dirección de correo electrónico para hacer este tipo de notificaciones es: etiquetado@aemps.es
- 4. Realizar seguimiento: sería recomendable que el farmacéutico ofreciera al paciente, afectado por la sospecha de reacción adversa, el Servicio de Seguimiento Farmacoterapéutico con el fin de evaluar su evolución.
- II. PROCEDIMIENTO DE DETECCIÓN Y NOTIFICACIÓN DE SOSPECHAS DE ENTRADA DE MEDICAMENTOS FALSIFICADOS A LA CADENA LEGAL DE SUMINISTRO
Ante la sospecha de entrada en la cadena legal de suministro de un medicamento falsificado, el farmacéutico debe:
- 1. Comprobar la procedencia del medicamento: el farmacéutico deberá verificar siempre que los proveedores, laboratorios o almacenes de distribución con los que trabaja cumplen con todos los requerimientos legales exigibles para el suministro y distribución de medicamentos. Para ello se podrá consultar el registro público de laboratorios7 y el catálogo de entidades de distribución8 disponible en la página web de la Agencia Española de Medicamentos y Productos Sanitarios.
En el caso de medicamentos sospechosos entregados por un paciente y que hayan sido adquiridos a través de internet u otras vías de suministro no legales, el farmacéutico intentará recabar la mayor cantidad posible de información.
En España, la venta por correspondencia y por procedimientos telemáticos de medicamentos sujetos a prescripción está prohibida. En el caso de medicamentos no sujetos a prescripción, la dispensación se realizará por una oficina de farmacia autorizada, con la intervención del farmacéutico y previo asesoramiento personalizado. La dispensación de medicamentos de uso humano no sujetos a prescripción a través de sitios web debe realizarse con los mismos principios de actuación profesional que la dispensación que pueda realizarse en una farmacia.
Las farmacias legalmente autorizadas para la venta a distancia, a través de sitios web, de medicamentos de uso humano no sujetos a prescripción notificarán esta actividad a la administración y mostrarán un logotipo identificativo común para toda la Unión Europea. El logotipo tendrá un enlace a la web de la AEMPS desde donde se podrá comprobar si efectivamente la farmacia cuenta con autorización para realizar esta actividad.
Cuando el comprador se encuentre en otro Estado Miembro, la venta a distancia a través de sitios web deberá realizarse de acuerdo a los requisitos establecimos en el RD 870/2013, de 8 de noviembre, por el que se regula la venta a distancia al público, a través de sitios web, de medicamentos de uso humano no sujetos a prescripción médica, así como los exigibles en el país de destino, tanto en relación a los medicamentos (incluyendo su etiquetado, prospecto y clasificación), como a las condiciones de dispensación.
7https://labofar.aemps.es/labofar/registro/farmaceutico/consulta.do?metodo=detalleBusqueda
8https://labofar.aemps.es/labofar/registro/entidadesDistribucion/consulta.do?metodo=detalleBusqueda
En el caso de detectar que una determinada página web está realizando presuntas actividades ilícitas de venta de medicamentos ilegales o falsificados, o sin la autorización correspondiente, el farmacéutico debería notificar esta sospecha a través de la Organización Farmacéutica Colegial.
El farmacéutico deberá ayudar a concienciar a la población sobre los riesgos de la compra de medicamentos en establecimientos no autorizados o a través de internet.
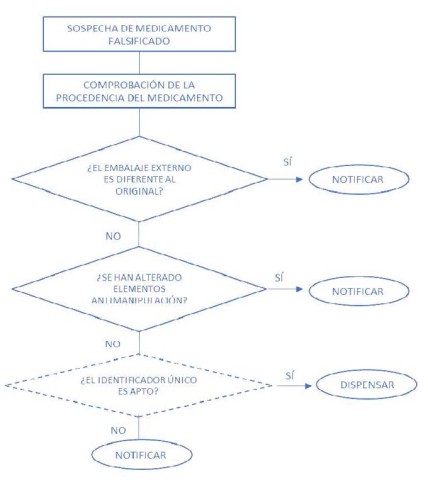
- Verificar el aspecto del embalaje externo, la existencia y el estado de los posibles dispositivos de seguridad del envase: en la actualidad algunos medicamentos ya incluyen elementos de seguridad como pueden ser sellos, hologramas, dispositivos para evitar la manipulación (apertura por troquelado, sellos, etc.), etc. Estos elementos serán cada vez más habituales junto con la incorporación de un número de identificación único que va a permitir la identificación individual de cada envase legal puesto en el mercado.
En la farmacia se comprobará, en el momento de la recepción del pedido, que tanto el aspecto, como los elementos de seguridad que pueda incluir un medicamento, están intactos y, cuando así esté disponible, se realizará la autenticación del envase antes de su dispensación a través de los procedimientos y la vía establecida.
- 3. Notificación de la sospecha de medicamento falsificado: el farmacéutico informará inmediatamente a la AEMPS de la sospecha de medicamento falsificado que haya detectado. La notificación se realizará a través de la dirección de correo electrónico habilitada para tal fin es medicamentfalsificados@aemps.es
La notificación podrán realizarla, además de farmacéuticos, otros profesionales sanitarios, titulares de la autorización de comercialización, laboratorios fabricantes/importadores y almacenes de distribución de medicamentos.
Esta dirección permite la notificación de sospechas de medicamentos falsificados detectados dentro de la cadena legal de suministro.
En la notificación se incluirá la siguiente información:
₋ Nombre del medicamento, forma farmacéutica y Código Nacional.
₋ Número de lote y fecha de caducidad.
₋ Lugar de detección o adquisición de la posible falsificación.
₋ Explicación de la sospecha y diferencias observadas entre la falsificación y el medicamento original.
₋ Otros datos de interés, fotografías.
₋ Nombre, apellidos, teléfono y correo electrónico de la persona que realiza la notificación.
El farmacéutico conservará el medicamento sospechoso ante posibles consultas e investigaciones por parte de la AEMPS.
La AEMPS se encargará de notificar a las Comunidades Autónomas, organizaciones profesionales, asociaciones de mayoristas y agentes implicados en la cadena de suministro de medicamentos, cualquier alerta sobre medicamentos falsificados detectados.
Buenas Practicas en Farmacia Comunitaria en Espana
CONSEJO GENERAL DE COLEGIOS OFICIALES DE FARMACEUTlCOS
DIAGRAMA DE FLUJO DEL PROCEDIMIENTO DE NOTIFICACION9 DE SOSPECHAS DE REACCIONES ADVERSAS A MEDICAMENTOS POR PARTE DEL FARMACEUTICO COMUNITARIO
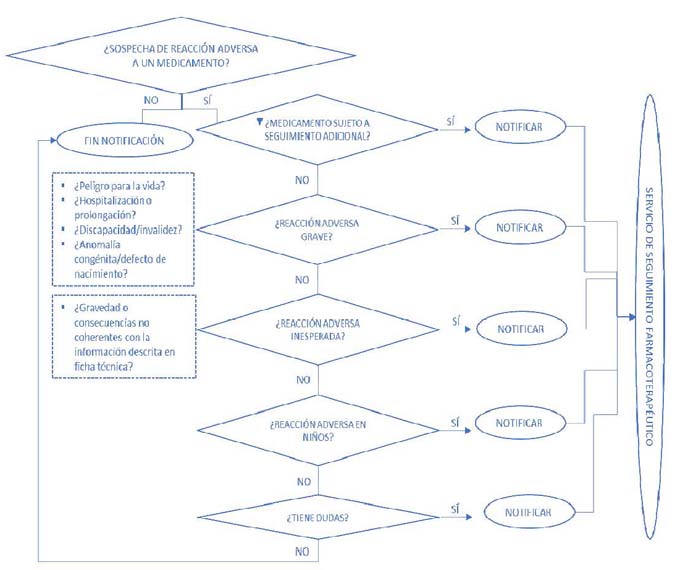
DIAGRAMA DE FLUJO DEL PROCEDIMIENTO DE DETECCIÓN Y NOTIFICACIÓN10 DE SOSPECHAS DE ENTRADA DE MEDICAMENTOS FALSIFICADOS A LA CADENA LEGAL DE SUMINISTRO
10 medicamentos.falsificados@aemps.e
INFORMACIÓN
- Erstad BL, Brophy GM, Martin SJ, et al. Key articles and guidelines relative to intensive care unit pharmacotherapy. Pharmacotherapy. 2009;29(10):1228-69
- Jefatura del Estado. Ley 10/2013, de 24 de julio, por la que se incorporan al ordenamiento jurídico español las Directivas 2010/84/UE del Parlamento Europeo y del Consejo de 15 de diciembre de 2010, sobre farmacovigilancia y 2011/62/UE del Parlamento Europeo y del Consejo de 8 de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la cadena de suministro legal, y se modifica la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. BOE-A-2013-8083. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/profesionales/legislacion/recopilacion/Paginas/in dicegeneral.aspx
- Jefatura del Estado. Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. BOE-A-2006-13554. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/legislacion/recopilacion/Paginas/leygarantiasyusoraciona.aspx
- Jefatura del Estado. Ley 44/2003, de 21 de noviembre, de ordenación e las profesiones sanitarias. BOE-A-2003-21340. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/legislacion/recopilacion/Paginas/leyordenacion.aspx
- Ministerio de Sanidad y Consumo. Real Decreto 1345/2007, de 11 de octubre, por el que se regula el procedimiento de autorización, registro y condiciones de dispensación de los medicamentos de uso humano fabricados industrialmente. BOE-A-A2007-19249. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/legislacion/recopilacion/Paginas/es pecialidadesfarmaceuticas.aspx
- Ministerio de Sanidad, Servicios Sociales e Igualdad. Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano. BOE-A-2013-8191. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/medicamentos/farmacovigilancia/Paginas/farmacovigilancia.aspx
- Ministerio de Sanidad, Servicios Sociales e Igualdad. Real Decreto 870/2013, de 8 de noviembre, por el que se regula la venta a distancia al público, a través de sitios web, de medicamentos de uso humano no sujetos a prescripción médica. BOE-A-2013-11728. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/medicamentos/medicamentos- internet/dispensacion-medicamentos-online/Paginas/Dispensacion- medicamentos-online.aspx
- Ministerio de Sanidad, Servicios Sociales e Igualdad. Estrategia frente a medicamentos falsificados 2012-2015. Agencia Española de Medicamentos y Productos Sanitarios. 2012. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.aemps.gob.es/publicaciones/publica/docs/Estrategia_falsificados_2012-2015.pdf
- Parlamento Europeo y Consejo de la Unión Europea. Directiva 2011/62/UE del Parlamento Europeo y del Consejo de 8 de junio de 2011 que modifica la Directica 2001/83/CE por la que se establece un código comunitario sobre medicamentos de uso humano, en lo relativo a la prevención de la entrada de medicamentos falsificados en la cadena de suministro legal. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://ec.europa.eu/health/files/eudralex/vol-1/dir_2011_62/dir_2011_62_es.pdf
- Varios autores. Documento de Buenas Prácticas en Farmacia Comunitaria en España. Consejo General de Colegios Oficiales de Farmacéuticos. 2013. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.portalfarma.com/Profesionales/Buenas-practicas- profesionales/Paginas/Buenas-practicas-Farmacia-Comunitaria.aspx
- World Health Organisation. Medicines: spurious/falsely- labelled/falsified/counterfeit (SFFC) medicines. WHO Factsheet Nº 275. 2012. [Internet – consultado el 24 de junio de 2014]. Disponible en: http://www.who.int/mediacentre/factsheets/fs275/en/







 Lilisbeth Perestelo:
Lilisbeth Perestelo: 


