
La Agencia Española de Medicamentos (Aemps) está llamada a jugar un papel aún más relevante en la evaluación de medicamentos en nuestro país. A las obligaciones propias como regulador, se le añadirán las que tenga que asumir por la aplicación del Reglamento de Evaluación de Tecnologías Sanitarias y es previsible que tenga que sumar otras nuevas que le otorgue el real decreto que se está preparando en el mismo ámbito. Diariofarma ha mantenido una conversación con la directora de la agencia, María Jesús Lamas, en la que expone su visión sobre la evaluación de tecnologías, algunos de los desarrollos normativos y leyes en marcha, tanto a nivel nacional como europeo, así como cuestiones relativas a los retos de la agencia, especialmente en materia de recursos humanos y actualización científica o los desabastecimientos. Estos últimos asuntos se publican en una pieza separada.
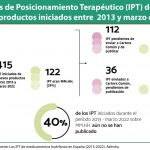
Pregunta. En julio del año pasado hubo una sentencia que anuló REvalMed y la Agencia reaccionó rápidamente y tomó las riendas de la elaboración de los informes de posicionamiento terapéutico (IPT). Desde fuera sorprendió un poco ya que estaba todavía pendiente la posibilidad de un recurso por parte del Ministerio, pero la Aemps publicó rápidamente un conjunto numeroso de IPT. ¿Cómo fue posible hacerlo así y por qué?
Respuesta. A nosotros no nos pilló desprevenidos. Ya teníamos un equipo preparado, trabajando en la elaboración de IPT. Cuando algo se hace de forma colaborativa entre voces distinta y con diferentes manos, como ocurría en Revalmed, lleva más tiempo que cuando lo hace un único equipo cohesionado y lo hace más eficientemente.
P. ¿Y cómo valora la sentencia?
R. La sentencia de la Audiencia Nacional era inequívoca. Nuestra asesoría jurídica nos dejó clarísimo que no había ninguna posibilidad de recurso. Había muchos IPT que estaban ya en marcha y se pudieron finalizar. Pudo parecer una aceleración, pero lo que hicimos fue acabar lo que estaba iniciado. Nuestra participación en iniciativas europeas, como EUnetHTA o los grupos de trabajo de los actos de implementación del reglamento, así como el conocimiento interno nos facilitaron la adaptación a esta nueva situación. Además, seguimos el espíritu de la sentencia ya que la Agencia es la única que tiene mandato y base legal para el posicionamiento terapéutico. Es discutible lo que establece sobre el análisis farmacoeconómico, pero sabiendo que lo que íbamos a hacer era provisional hasta que se aprobara el nuevo modelo, decidimos seguir adelante.
______
“Hay que asegurar que las cuestiones medio ambientales no ponen en riesgo la disponibilidad de medicamentos necesarios para tratar a los pacientes”
______
P. Ha comentado que la pandemia hizo establecer lazos entre los trabajadores de la Aemps. Una de esas personas era César Hernández, antes jefe del Departamento de Medicamentos de Uso Humano en la agencia y ahora director general de Cartera de Servicios del SNS y Farmacia del Ministerio. ¿Esa relación facilitó el entendimiento en ese momento?
R. La relación con César Hernández era excelente cuando estaba en la Agencia y lo sigue siendo. Tuvo una contribución valiosísima en la lucha contra la pandemia gracias a su buen criterio. No siempre estamos de acuerdo, como ocurre con las personas con buen criterio, pero esto lo hablamos con él y la secretaria de Estado del momento. Lo más importante es que hay un plan para sacar el real decreto de evaluación, donde se debe definir bien el objeto, metodología, gobernanza y participación.
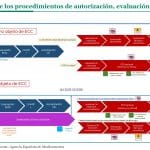
P. Luego entramos en el real decreto, pero, en primer lugar, respecto del reglamento: dado que la afectación será secuencial, según los tipos de medicamentos, ¿eso complicará a la Aemps, que tendrá por un lado que gestionar los joint clinical assessments (JCA), al tiempo que los IPT nacionales?
R. Desde el inicio de esta nueva etapa, en julio pasado, hemos trabajado para asegurarnos de que nuestro modelo de evaluación fuera perfectamente encajable con el del reglamento europeo, a medida que se aplique, primero para medicamentos oncológicos y terapias avanzadas, en enero de 2025, luego enfermedades raras en 2028, y el resto en 2030.
______
“Tras la sentencia de Revalmed, seguimos su espíritu ya que la Agencia es la única que tiene mandato y base legal para el posicionamiento terapéutico”
______
P. ¿Cómo debería ser el modelo de evaluación?
R. Creemos que debe haber distintos niveles de actuación y agentes. Primero una evaluación científico-técnica, que tiene como objetivo determinar el valor clínico añadido de las nuevas tecnologías sanitarias. Es una evaluación de eficacia relativa y lo que se extrae es el valor en eficacia y seguridad comparado con las alternativas disponibles. Esta parte, puramente técnica es el objeto de lo que la Comisión Europea llama JCA. También se podría hacer una evaluación económica también puramente científica. Posteriormente a esto hay que tener en cuenta otras cuestiones, se trata de variables como los aspectos éticos, sociales u organizativos, que deben ser tenidos en cuenta y eso es el posicionamiento de la tecnología. Aquí deberían tener voz los pacientes; los profesionales, las comunidades autónomas, como pagadores y responsables de la planificación y organización sanitaria. Posteriormente, la decisión política de precio y reembolso que se toma en la Comisión Interministerial de Precios de los Medicamentos (CIPM). Creo que es higiénico separar estos niveles para evitar potenciales conflictos de interés.
P. ¿Y cómo adaptarán las JCA?
R. La JCA ni puede tener conclusiones o valoraciones subjetivas, pero nosotros estamos trabajando en definir las conclusiones sobre el beneficio clínico añadido. Por ejemplo: es una opción más; es una alternativa equivalente; tiene beneficio clínico añadido. El grado también dependerá de la magnitud del beneficio y la calidad de la evidencia en que se sostiene el análisis. Estamos trabajando en análisis internos con los IPT elaborados hasta ahora para ver si esta metodología a aplicar se reproduce en los informes que vamos sacando para categorizar el nivel de beneficio clínico. Es una conclusión técnica y no un posicionamiento, que debe tener en cuenta otras consideraciones.
P. ¿La Aemps dispone de los profesionales formados para realizar las evaluaciones económicas dentro de este marco?
R. Los profesionales capacitados en evaluaciones económicas existen, pero aún no forman parte de la Agencia. El futuro Real Decreto de evaluación de tecnologías sanitarias definirá quién realiza cada aspecto de la evaluación. Y si la evaluación económica recae en la misma unidad que la evaluación clínica, habrá que dotarla de los recursos necesarios para hacerlo.
______
“Nuestra aproximación se centra en exigir una rigurosa evidencia científica y un seguimiento detallado de los pacientes que participan en los programas de autorización de uso”
______
P. Es una propuesta que me recuerda a la de Funcas sobre la creación de una agencia de evaluación a dos velocidades. Inicialmente insertada dentro de la Aemps y, posteriormente, realizar una spin off
R. Se puede parecer en algunas cuestiones, como la visión de separar la evaluación técnica y científica de otras consideraciones en el proceso de incorporación de tecnologías sanitarias. Coincidimos en este enfoque basado en una metodología transparente, con una gobernanza establecida y participativa.
P. Pasando a otros temas sobre normativa. ¿En qué situación está el real decreto de Medicamentos en Situaciones Especiales, del que se hizo una audiencia pública previa en 2020…?
R. Está escrito, pero a la espera de otros desarrollos legislativos complementarios. Actualmente se contempla como situación especial los medicamentos autorizados en espera de decisión de precio y financiación, pero esto es una irregularidad. En 2009 no se pensó en esa situación, ya que los usos especiales eran medicamentos extranjeros, uso compasivo o fuera de ficha técnica. En este último ya está claro que la Aemps no debe intervenir, sino que es un tema de práctica clínica. Si un medicamento está autorizado por la Comisión Europea, estrictamente, no es extranjero ya que hay un mercado único y tampoco es uso compasivo. Se considera extranjero porque hay una necesidad de los pacientes, pero esta situación no es buena ni cómoda para nadie y menos si se alarga. Perjudica las negociaciones y obliga a las comunidades autónomas a pagar por un medicamento que aún no tiene resolución de financiación. Yo llegué en el 2018 y desde el 19 tenemos planificado acometer esto. Desde la autorización por la CE hasta el precio-reembolso es un periodo que hasta ahora no está regulado, pero quien tendría que decidir serían las comunidades autónomas, estos medicamentos no deberían ser objeto de análisis por parte de la Aemps. Aunque lo hacemos de manera informal, no me parece adecuado. Es necesario aclarar esto y necesitamos el real decreto de financiación y precio y la actualización de la Ley de Garantías, ya que, si no, solo regularíamos el uso compasivo y los medicamentos extranjeros.

P. El paquete farmacéutico que ha finalizado su tramitación en el Parlamento recoge, entre otras, una cuestión relevante, como la exención hospitalaria, donde España ha sido importante al aportar su experiencia. ¿Cómo lo valora?
R. La propuesta del paquete farmacéutico, inspirada en la experiencia española, es vista positivamente. Antes de la propuesta de la Comisión en muchos comités técnicos se habló de este asunto. El modelo español, que al principio generaba recelos, creo que finalmente ha sido abrazado por diferentes países y la Comisión. La exención hospitalaria garantiza la generación de evidencia científica suficiente para proteger a los pacientes y al tiempo, para asegurarnos de que si puede ser utilizada de forma relativamente amplia pueda llegar a ser un medicamento autorizado. Nosotros distinguimos entre dos tipos terapias avanzadas susceptibles de ser autorizadas como exención hospitalaria. Por un lado, las que se dirigen a nichos muy pequeños de pacientes, que pueden no ser de interés comercial, y por otro lado, aquellas con mayor potencial. El objetivo es que estos últimos, incluso cuando provienen del ámbito académico o de hospitales, puedan eventualmente alcanzar una autorización centralizada, beneficiando a un mayor número de pacientes. Esto es lo que está haciendo el Hospital Clinic con el CDTI para el ARI-001. Se nos puede decir que esta vía regulatoria es costosa y difícil, pero eso no puede ser lo que impida que lleguen a todos los pacientes. Nuestro enfoque no es disminuir las exigencias científicas sino facilitar el camino para que estas terapias lleguen a más pacientes. Por eso, la propuesta incluye asesorías y acceso a la vía PRIME de la EMA para apoyar a pequeñas y medianas empresas, así como a grupos académicos, en el desarrollo de estas terapias. Nos gusta el modelo y creemos que es bueno para la ciencia, el conocimiento, los pacientes, los investigadores, el sistema…
P. Entonces, ¿cree que todas las terapias avanzadas deberían esforzarse por alcanzar una autorización centralizada, también para otorgar a los pacientes las mismas garantías que en el resto de los medicamentos?
R. Sí, especialmente aquellas terapias que tienen el potencial de beneficiar a un número significativo de pacientes. Debería haber también un registro que indique los lugares donde se están fabricando y administrando y, por otro lado, dar el soporte regulatorio posible para hacerlo realidad. Puede ocurrir que una gran compañía compre el producto y desarrolle el proceso regulatorio, pero este no es el modelo deseable de España, donde queremos que haya participación pública y que se beneficie todo el SNS.
______
“Si un medicamento está autorizado por la Comisión Europea, estrictamente no es extranjero ya que hay un mercado único y tampoco es uso compasivo”
______
P. En cuanto al uso de la exención hospitalaria ha habido mucho debate en relación con poner límites temporales o de número de pacientes antes de que tengan que solicitar una licencia. ¿Cómo lo ve?
R. No creo que sería necesario ya que perjudicaría a la investigación más que beneficiarla. Nuestra aproximación se centra en exigir una rigurosa evidencia científica y un seguimiento detallado de los pacientes que participan en los programas de autorización de uso. La farmacovigilancia se evalúa con los mismos estándares exigidos para cualquier medicamento industrial.
P. Las garantías son las mismas…
R. Son las mismas
P. En el caso del ARI-001, se han presentado datos de uso en indicaciones diferentes a las autorizadas en la exención de uso. ¿Eso son usos compasivos que se han ido autorizando?
R. Si, y ensayos clínicos creo recordar. Imagina que en vez de ser un medicamento con clausula de exención hospitalaria y un hospital es un medicamento de desarrollo industrial y una empresa farmacéutica. Es totalmente factible que se diseñen terapias basadas en la misma tecnología, contra las mismas dianas y que se presenten datos propios en congresos. La clave es que cada indicación debe estar sustentada por los datos generados por quienes lideran el desarrollo, sin depender de la evidencia generada por productos industriales similares. Si son propietarios de su desarrollo, ¿por qué no presentar esos datos y utilizarlos?
P. En el paquete farmacéutico se plantean algunas cuestiones medioambientales que pueden poner en riesgo la investigación, desarrollo y fabricación de algunos medicamentos. ¿Cómo lo valora?
R. Este es un tema de gran complejidad y preocupación. Aún no se ha discutido en detalle este asunto en el Consejo. Lo que cambia es que un medicamento podría no ser autorizado de acuerdo con la evaluación de impacto medioambiental. Veremos cual es el resultado de las negociaciones.
______
“Hemos trabajado para asegurarnos de que nuestro modelo de evaluación sea perfectamente encajable con el del reglamento europeo de las JCA”
______
P. Puede afectar incluso a medicamentos antiguos, algunos de ellos, esenciales…
R. Sí, a los anteriores a 2005. Hay que asegurar que todas estas medidas no ponen en riesgo la disponibilidad de medicamentos necesarios para tratar a los pacientes. Al mismo tiempo, es importante velar por una industria farmacéutica sostenible. Y esto va ligado a otras cosas, como cuando hablamos de la necesidad de que Europa tenga una autonomía estratégica abierta. Hay voces abogando por una relocalización de la industria farmacéutica, pero esto no se hace a cualquier precio ya que si Europa marca estándares al resto del mundo, debe ser capaz de desarrollas una industria farmacéutica compatible con le medio ambiente.
P. ¿Podría la exigencia de estándares medioambientales para medicamentos fabricados en Europa poner a nuestra industria en situación de desventaja con respecto de los fabricados fuera?
R. Está todo en discusión. Hay voces que dicen que se deberían exigir los mismos requisitos para la fabricación en terceros países. Es complejo, pero la deslocalización no es algo exclusivo de esta industria, tanto otras industria innovadoras o no han deslocalizado su producción y los estándares laborales o medioambientales no son los mismos. Hay que ver hasta dónde realizar esas exigencias. Habrá que determinar cómo queremos fabricar nosotros en Europa y luego seguir con que las relaciones comerciales se hagan con socios fiables en el sentido más amplio de la palabra.
Fotos: Irene Medina.














 César Hernández, director general de Cartera y Farmacia del Ministerio de Sanidad:
César Hernández, director general de Cartera y Farmacia del Ministerio de Sanidad:  Kilian Sánchez, secretario de Sanidad del PSOE y portavoz de la Comisión de Sanidad del Senado.:
Kilian Sánchez, secretario de Sanidad del PSOE y portavoz de la Comisión de Sanidad del Senado.:  Rocío Hernández, consejera de Salud de Andalucía:
Rocío Hernández, consejera de Salud de Andalucía:  Nicolás González Casares, eurodiputado de Socialistas & Demócratas (S&D - PSOE):
Nicolás González Casares, eurodiputado de Socialistas & Demócratas (S&D - PSOE):  Juan José Pedreño, consejero de Salud de Murcia:
Juan José Pedreño, consejero de Salud de Murcia: